Fluorescence Polarization in Life Sciences
1. Introduction
The application of fluorescence polarization offers unique advantages over conventional fluorescence imaging and quantitation. In fluorescence polarization studies, fluorophores tied to biological samples or chemical compounds help elucidate the underlying mechanism of interest. As opposed to the standard fluorescence techniques, fluorescence polarization allows for fast and accurate quantitative measurements with relatively simple instrumentation (compared to sophisticated research instruments). Therefore, applications such as High Throughput Screening (HTS), a key technique in drug discovery, have made widespread use of fluorescence polarization. Progressively more complicated studies at the molecular level are being conducted by the application of microscopy-based fluorescence polarization studies. Excellent technical information on the theory and applications of fluorescence polarization is available in the literature [1-6]. However many of these resources provide a limited perspective on applications. This article is a concise review of various applications in life sciences that are based on fluorescence polarization.
2. The Fundamentals
Polarization is a fundamental property of light [7]. Light is considered to be linearly polarized when the orientation of its electric field vector does not change during propagation. Fluorescence polarization studies utilize linearly polarized light for the illumination of the sample.
The interaction of polarized light with a fluorophore can be best described by considering the concept of an electric dipole. A dipole refers to the formation of separated positive and negative charges and forms the basis of a simple classical model of the interaction of light with matter. The dipole moment (a vector) denotes the strength of the dipole and its direction. Fluorophores tend to have a dipole moment, and the excitation dipole moment of a fluorophore can be different from its emission dipole moment. The dipole moment of a fluorophore is also influenced by the environmental factors. When the electric field of the excitation light is parallel to a fluorophore’s absorption dipole moment the fluorophore has the greachance of absorbing a photon, which leads to its preferential excitation among the population of illuminated fluorophores. On the other hand, when the absorption dipole moment is perpendicular to the electric field of excitation then the fluorophore cannot be excited. Furthermore, when the excited fluorophore reemits light, the emission is polarized parallel to the plane of its dipole. These phenomena are the basis for fluorescence polarization studies. They occur in all fluorescence applications at the molecular level, but are generally not apparent in conventional fluorescence studies. Due to the random orientations of individual fluorophores in a sample that is typically excited by unpolarized light, the polarization effect is lost due to averaging.
3. Principle of Application
Figure 1 illustrates the basic principle employed in conventional fluorescence polarization studies. A fluorescently labeled sample is illuminated with linearly polarized light at the wavelength of the fluorophore absorption and the longer wavelength emission from the sample passes through a rotatable linearly polarizing filter (analyzer) before detection. Since the degree of polarization cannot be detected directly by conventional detectors such as photomultiplier tubes (PMTs), an observation of the change in the fluorescence intensity is used to indirectly study polarization.
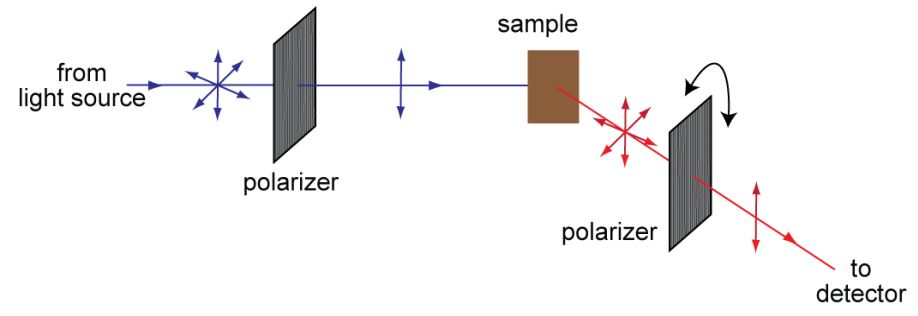
Figure 1: Fluorescence polarization measurement. A monochromatic beam illuminates the fluorophores within the sample. A linear polarizer located in the illumination beam attenuates the randomly oriented polarization states except for those in one plane, thereby generating linearly polarized light. This plane of polarized light also becomes the reference plane. Depending upon the orientation of their absorption dipoles, individual fluorophore molecules are preferentially excited (see Section 2). The emission from the fluorescent sample can be considered as another light source, composed of the combination of signals from the emission dipoles of individual fluorophore molecules. If the absorption and emission dipole moments of all the fluorophores were to be aligned with the electric field vector of the illumination beam (plane polarized light) then the emission signal would also be plane polarized. However, this is typically not the case. Therefore, the emission signal is partially depolarized due to the random orientation of the fluorophores, even if the fluorophores are completely static. As the molecules move during the time window of detection, the emission becomes even more depolarized. A polarizer placed in front of the detector is used to detect the intensity of emission light in a given plane of polarization. The intensity measurement made with the emission polarizer oriented orthogonal to the electric field of illumination beam (the reference plane) is denoted as I⊥ and when the emission polarizer is rotated by 90 degrees with respect to the I⊥|| measurement is made.
A ratio derived using two different fluorescence intensity measurements (see Fig. 1) provides a measure of the degree of polarization of the emission. Two expressions that are used interchangeably to study polarization are “polarization ratio” ( p ) and “emission anisotropy” ( r ). These dimensionless quantities are calculated as follows [1]:
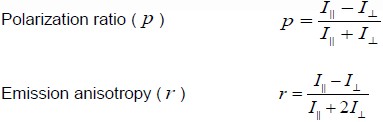
where I|| and I⊥ are the intensity measurements of the emission signal made parallel to or orthogonal to, respectively, the direction of the electric field of illumination light (determined by the polarizer in the excitation light path). Note that I || and I⊥ measurements can also be made by rotating the polarizer in the excitation light path instead of rotating the emission polarizer. If all of the emission dipoles were to reradiate light parallel to the electric field of the illumination light each absorbed, p would equal 1 (and r would also equal 1), whereas if all the emission dipoles were to reradiate light perpendicular to the illumination light electric field then p would be -1 (and r would be -0.5). Therefore in a properly calibrated experiment, the values of p and r can vary within these limits. In particular, p and r do not reach their limiting values because the dipole moments of the fluorophore molecules are not completely aligned in any one direction.
The relationship between p and r is given by r = 2 p /(3 − p) or p = 3r /(2 + r) . Regarding the use of p versus r as the measured value of polarization, many biological applications use emission anisotropy ( r ) because it offers advantages over polarization ratio ( p ) in the physical interpretation of measured data. However, historically, polarization ratio ( p ) has been used in the clinical chemistry and drug discovery. One of the advantages of polarization studies is that since the measurements are ratiometric in nature they are not dependent on the actual light intensity values or the fluorophore concentration.
What is the significance of relative strength of I|| and I⊥? The majority of FP studies are based on the premise that rotational diffusion of fluorophores is the dominant cause of fluorescence depolarization (a change in the degree of polarization). Depolarization can be caused by several factors such as the optical system itself used for polarization measurement, or fluorescence resonance energy transfer (FRET), or a change in the orientation of the dipole between excitation and emission events. Note that (partial) depolarization is intrinsically present in all fluorescence polarization applications due to the random orientation of the population of fluorophores that behave as spatially incoherent light sources (see Fig.1). Therefore, in real life experiments I|| and I⊥ are only relative measurements that refer to the maximum or minimum signals.
Fluorescence polarization was first described by Perrin [8] and is elucidated by the following mathematical equation called the Perrin equation:
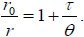
In this equation τ is the fluorescence lifetime (the average time a fluorophore stays in the excited state before emitting a photon), θ is a rotational correlation time, r is the measured anisotropy andr 0 is the fundamental anisotropy (in the absence of any rotational diffusion). Also note that the rotational correlation time of the fluorophore (θ) can be described as [2]:
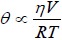
where η is the viscosity of the sample, R is the gas constant, T is absolute temperature, and V is the volume of the rotating unit (i.e. the fluorescently labeled molecule). This relation implies that molecular size affects the polarization of fluorescence emission. Therefore slowly tumbling molecules in solution generate a higher r (or p ) value compared to molecules that are tumbling faster. However when θ <<τ , the orientations of individual fluorophores are rerandomized by the time measurements are made and the net emission is unpolarized ( r →0 ). And when θ >>τ then immediately following excitation the measured anisotropy approaches fundamental anisotropy (r → r0).
4. Applications of Fluorescence Polarization
The measurement of fluorescence polarization (or anisotropy) adds another dimension of observation to a fluorescence-based experiment. This added dimension can provide information on the local environment, fluorescence lifetime and molecular mass. A variety of instruments are utilized in fluorescence polarization studies. These instruments are based on the design of existing fluorescence spectroscopy or microscopy techniques. Depending upon the requirements of an application a suitable instrument and a compatible assay may be selected.
Spectroscopy based fluorescence polarization studies
Traditionally spectroscopic instruments such as a fluorometer (see Fig. 2) have been used for fluorescence polarization studies. In these instruments the sample is placed in a cuvette or a tube. As shown in this figure, the excitation and emission wavelengths of light are selected using monochromators. The signal intensity from the fluorophores is recorded on a nonpixelated detector such as a PMT. This allows for the measurement of bulk properties of a population of fluorophores.
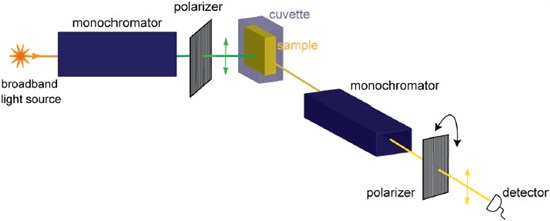
Figure 2: Spectrofluorometer for polarization measurement. One of the polarizers (typically the polarizer in the emission light path) is rotated along parallel or orthogonal directions to obtain I|| and I⊥ measurements. This is an L-format (or single channel) spectrofluorometer. In a T-format (or two channel) spectrofluorometer (see the principle in the emission light path of Figs. 3 & 4B) the emission signal from the sample is split into two channels, each of which is used to measure different polarization states, I||and I⊥ simultaneously.
This measurement approach has been in use for several decades for applications targeted towards biochemical research or clinical diagnostics. For example the measurement of fundamental anisotropy ( r0 ) and an understanding of the electronic properties of fluorophores can be obtained by anisotropy measurements at different wavelengths. Perrin plots (based upon the Perrin equations, above) of labeled macromolecules have been extensively used to determine the apparent hydrodynamic volumes [1]. Historically biological membranes have also been studied using fluorescence anisotropy measurements. Other biological applications include the study of association of proteins with larger molecules.
Fluorescence polarization studies for HTS
One of the bottlenecks in the application of the above measurement configuration in life sciences has been the use of a cuvette or a tube for sample preparation. For example, the sample preparation conventions of HTS require analysis in microplates. HTS plays a key role in the small-molecule drug discovery process. In this technique, a large number of synthetic compounds are tested against biological targets (such as a gene or a protein affecting a disease, for example) in in vitro assays. The objective of this screening is to identify a lead, or a promising molecule that can interact with the target and hence has potential therapeutic application. Since it might be necessary to screen millions of compounds against the target, the time required for each assay is crucial. In this regard an advantage of fluorescence polarization assays is that they are homogeneous; i.e., these assays do not require separation of bound and free species, thereby making it easier to automate the experiments so that results can be obtained almost instantaneously and hence enhance the overall throughput. This reason has led to significant advances in the application of fluorescence polarization assays in HTS.
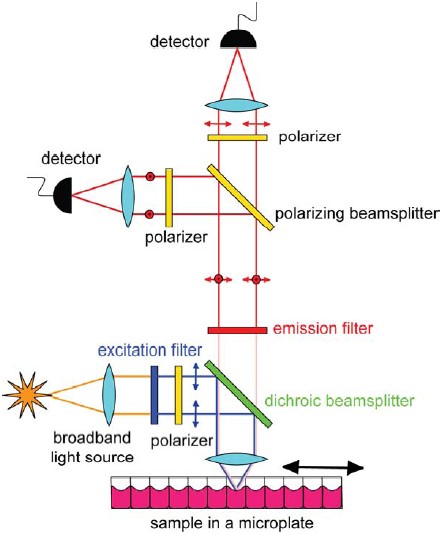
Figure 3: A modern HTS instrument essentially employs the principle of operation of a conventional fluorescence microscope. Typically excitation and emission filters are used to select monochromatic wavelengths of light in the excitation and emission channel. The emission signal is split into two channels each of which is utilized to simultaneously measure I|| and I⊥ components using a non-pixelated detector such as a PMT. The sample is placed in microwells that have 96, 384 or 1536 format. This allows for screening of a large number of compounds in a given experiment.
Figure 3 shows the principle of operation of a modern HTS instrument that utilizes optical filters for distinguishing excitation light from emission signal. Plane polarized excitation light is used to illuminate a fluorescently labeled sample and the emission from the sample is split into two channels, each of which is used to measure different polarization states, (i.e. || and I ⊥ intensities). Simultaneous acquisition of two orthogonal polarization components has the advantage of minimizing artifacts resulting from photobleaching and movement of the sample, and is also faster than two sequential measurements (see Figs.1 & 2). Depending upon the flexibility of requirements, monochromators may be used in the illumination and emission paths. Monochromators provide greater flexibility in working with different wavelengths and therefore with different colored fluorophores. However the use of optical filters has the advantage of providing higher blocking of excitation light into the emission channel and therefore higher signal-to-noise ratio leading to enhanced throughput.
The first generation HTS instruments launched in mid 90’s enabled significant throughput advancement in the drug development process [2-3]. In order to further optimize the cost of screening, the assays needed to be miniaturized, leading to the development of second generation HTS instruments that provide much higher throughput and can handle a larger number of wells (384 or even 1536, compared to the original standard of 96 wells) in an automated fashion.
HTS fluorescence polarization assays are generally performed under equilibrium conditions. For example, the affinity of a protein-DNA interaction can be determined by monitoring the steady-state (equilibrium condition) fluorescence anisotropy changes associated with the formation of the complex. An example of a direct binding assay that is commonly employed in HTS might look like [2, 6]:
where F refers to the fluorophore that is covalently bound to the Ligand forming a F-Ligand complex. This complex and the Receptor have a non-covalent interaction. Owing to a significant difference in their sizes, under equilibrium condition, the emission signal from the F-Ligand complex has much lower polarization value compared to that of the F-Ligand:Receptor.
However, the most popular HTS assay utilizing fluorescence polarization is the competition binding assay:
In this assay, I is an inhibitor (for example, another ligand which is not fluorescently labeled) that competes with the F-Ligand complex in binding with the Receptor. In these experiments the affinity of binding of F-Ligand with that of the Receptor is known and therefore this technique enables determination of the binding affinity of a ligand without fluorescently labeling it, thus simplifying the sample preparation step. Some of the classes of targets that utilize this technique are protein-protein interaction, GPCR (G-protein coupled receptors that are involved in signaling pathways across cellular membranes), nuclear receptors, protein kinase, and phosphatase.
Other types of assay modes for HTS include enzyme assays (direct or competitive immunodetection, transferase) and protease assays (based on size reduction or detection by protein binding). The applications of these assays include receptor and ligand binding studies, protein-peptide interactions and DNA-protein interactions.
Fluorescence polarization experiments are not limited to only equilibrium binding studies but are also applicable to "real-time" experiments. Such experiments can measure fluorescence polarization as a function of time and therefore help understand the kinetics of the assay.
Microscopy based fluorescence polarization studies
Depending upon the requirements, fluorescence polarization assays can be performed in steady state or by utilizing time-resolved measurements (such as fluorescence lifetime imaging microscopy or FLIM). Such measurements can be done in the time or the frequency domain utilizing widefield or scanning methods (see Fig. 4). Since microscopy techniques utilize fairly sophisticated optics and instrumentation they allow for high sensitivity as well as high resolution imaging of a fluorescently labeled sample. For example, several microscopy techniques utilize high numerical aperture (NA) objectives which work with a high refractive index medium (typically oil) between the coverglass and the objective lens. While high NA objectives allow for better resolution, they can significantly alter the polarization state of the light and therefore require careful calibration. Additionally, the high cost of such instruments prohibits their widespread application, in contrast to HTS, and therefore they are primarily used for research applications.
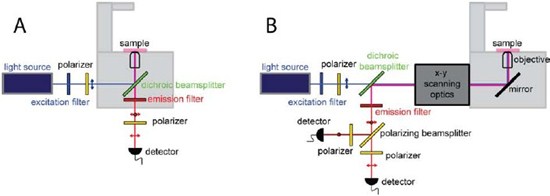
Figure 4: Microscopy based fluorescence polarization. (A) Schematic of a widefield imaging system. (B) Scanning instrument based fluorescence imaging. Depending upon the application, a pixelated or a nonpixelated detector such as an avalanche photodiode (APD) can be used to collect the emission signal. Additionally advanced instrumentation may be utilized for the measurement of fluorescence decay and lifetime imaging using the time-domain or the frequency-domain measurements.
We have already noted that fluorescence polarization studies uniquely allow the determination of the chemical and physical environments of macromolecules in intracellular environments. Even within a given fluorescence experiment, polarization measurement provides complimentary information to that of the original fluorescence experiment.
For example, Fluorescence Resonance Energy Transfer (FRET) is used to study molecular interactions at very small distances (generally less than 10 nm). Typically FRET refers to hetero-FRET, i.e., FRET occurring between two (or more) different types of fluorophores consisting of a donor-acceptor pair. Several approaches are available to study hetero-FRET that measure a change in the property (such as, lifetime or intensity) of the donor and acceptor. However, another type of FRET that occurs between the same species of a fluorophore, termed homo-FRET, can only be studied using fluorescence polarization measurements. Neither observed fluorescent intensity nor the lifetime of the fluorophore changes in the presence of homo-FRET. Fluorescence polarization allows identification of homo-FRET because fluorophores that are directly excited by polarized illumination light exhibit highly polarized emission signals compared to the sub-population of fluorophores that were excited by homo-FRET. Therefore in the absence of other depolarization factors, homo-FRET leads to a decrease in fluorescence anisotropy. An advantage in the study of homo-FRET is that a large spectral window is available (since only one type of fluorophore is used) therefore benefitting multiplexing assays. The study of homo-FRET using fluorescence polarization has been utilized for several investigations in biological environments. For example, homo-FRET studies have enabled determination of the oligomerization states of membrane proteins and the study of heterogeneity in lipid-order in the plasma membrane of cells. Such studies elucidate the complex associations of states of molecules and how they relate to biological functions [4].
Even hetero-FRET measurements using fluorescence polarization offer potentially higher dynamic range and temporal resolution [9-10] compared to conventional FRET measurement techniques. Another example is time-domain FLIM [4] combined with polarization measurement which is an important tool for the investigation of molecular rotation, binding reactions, and protein-protein interactions in living cells. FLIM based polarization studies remain one of the few methods that can differentiate among monomers, dimers, trimers and higher order assemblies in live cells [5]. However due to the complexity of implementation and analysis, time-resolved fluorescence anisotropy applications based on FLIM are still not widely prevalent.
Polarized Total Internal Reflection Fluorescence (polTIRF) microscopy (Fig. 4A) is another variant of an existing fluorescence microscopy technique (i.e. TIRF microscopy) that has been used to study the motility properties of motor proteins such as myosin along actin filaments. This technique even allows for the study of the orientation of individual molecules. With the illumination of the sample at various polarization angles it has become possible to understand the tilting and rotational motions of individual molecules [11].
Conclusion
Given the unique advantages offered by fluorescence polarization, it has found widespread application in disciplines such as in HTS. However, its full potential still remains untapped for research applications.
References
[1] Lakowicz, J.R., Principles of Fluorescence Spectroscopy, Springer, New York, USA, 1999.
[2] Owicki, J.C., Fluorescence Polarization and Anisotropy in High Throughput Screening: Perspectives and Primer, Journal of Biomolecular Screening, 5 (5), 297-306, 2000.
[3] Nasir, M.S., Jolley, M.E., Fluorescence Polarization: An Analytical Tool for Immunoassay and Drug Discovery, Combinatorial Chemistry & High Throughput Screening, 1999, 2, 177-190, 1999.
[4] Levitt, J.A., Matthews, D.R, Ameer-Beg, S.M. and Suhling, K., Fluorescence Lifetime and Polarization Resolved Imaging in Cell Biology, Current Opinion in Biotechnology, 20:28-36, 2009.
[5] Vogel, S.S., Thaler, C, Blank, P.S., Koushik, S., Time Resolved Fluorescence Anisotropy, Chapter 10, FLIM Microscopy in Biology and Medicine, CRC Press Taylor & Francis Group, USA, 2009.
[6] Fluorescence Polarization Technical Resource Guide (4th Edition), Invitrogen | ThermoFisher Scientific
[7] Erdogan, T., Understanding Polarization, IDEX Health & Science
[8] Perrin, M.F. Polarization de la luniere de fluorescence. Vie moyenne de molecules dans l’etat excite”, J. Phys. Radium, 7, 390-401, 1926.
[9] Mattheyses A.L., Hoppe A.D., Axelrod D., Polarized fluorescence resonance energy transfer microscopy, Biophysical Journal, 87(4):2787-97, 2004.
[10] Rizzo M.A., Piston D.W., High-contrast Imaging of Fluorescent Protein FRET by Fluorescence Polarization Microscopy, Biophysical Journal, 88(2):L14-6, 2005.
[11] Rosenberg, S.A., Quinlan, M.E., Forkey, J.N., and Goldman, Y.E., Rotational Motions of Macromolecules by Single-Molecule Fluorescence Microscopy, Accounts of Biochemical Research, 38(7), 583-593, 2005.
Authors
Prashant Prabhat, Ph.D. and Turan Erdogan, Ph.D.